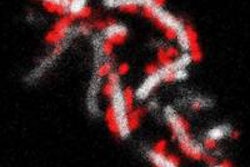
University of Pennsylvania (Penn) researchers have found that the periodontal bacterium Porphyromonas gingivalis acts on two molecular pathways to simultaneously block immune cells' killing ability while preserving the cells' ability to cause inflammation. Their findings are reported in the journal Cell Host & Microbe (June 11, 2014, Vol. 15:6, pp. 768-778).
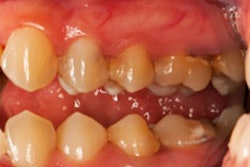
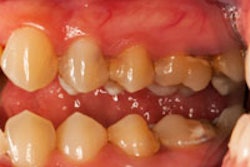
In the study, the researchers show that the bacteria responsible for many cases of periodontitis create an imbalance with a two-pronged attack of the human immune system. This imbalance is known as dysbiosis. This discovery opens up new targets for periodontitis treatment and also suggests a bacterial strategy in other diseases involving dysbiosis, they noted.
 George Hajishengallis, DDS, PhD, professor, University of Pennsylvania School of Dental Medicine Department of Microbiology. Image courtesy of the University of Pennsylvania.
George Hajishengallis, DDS, PhD, professor, University of Pennsylvania School of Dental Medicine Department of Microbiology. Image courtesy of the University of Pennsylvania.The idea, known in ecology as a keystone species, suggests that, although P. gingivalis may be relatively few in number in the mouth, their presence exerts a pull on the overall microbial ecosystem larger than its presence. Indeed, the team has shown that, although P. gingivalis is responsible for instigating the process that leads to periodontitis, it cannot cause the disease by itself.
What this means in the mouth is that so-called "bystander" gum bacteria aren't cleared by the immune system, promoting dysbiosis and leading to the bone loss and inflammation that characterizes periodontitis. At the same time, breakdown products produced by inflammation provide essential nutrients that "feed" the dysbiotic microbial community. The result is a vicious cycle in which inflammation and dysbiosis reinforce one another, exacerbating periodontitis.
Work by George Hajishengallis, DDS, PhD, a professor in the Penn School of Dental Medicine Department of Microbiology, and his group had previously identified P. gingivalis as a keystone pathogen.
"Scientists are beginning to suspect that keystone pathogens might be playing a role in irritable bowel disease, colon cancer, and other inflammatory diseases," lead study author Dr. Hajishengallis stated in a press release. "They're bugs that can't mediate the disease on their own; they need other, normally nonpathogenic bacteria to cause the inflammation."
In this study, Dr. Hajishengallis and his co-authors wanted to more fully understand the molecules involved in the process by which P. gingivalis caused disease.
"We asked the question, how could bacteria evade killing without shutting off inflammation, which they need to obtain their food," Dr. Hajishengallis said.
So the team focused on neutrophils, which are often the body's first responder to periodonal insults. Based on the findings of previous studies, the researchers examined the role of two protein receptors: C5aR and Toll-like receptor-2 (TLR2).
After inoculating mice with P. gingivalis, the researchers found that animals that lacked either of these receptors had lower levels of bacteria than untreated, normal mice. Blocking either of these receptors on human neutrophils in culture also significantly enhanced the cells' ability to kill the bacteria. Microscopy revealed that P. gingivalis causes TLR2 and C5aR to physically come together.
"These findings suggest that there is some crosstalk between TLR2 and C5aR," Dr. Hajishengallis said. "Without either one, the bacteria weren't as effective at colonizing the gums."
Further experiments in mice and cultured human neutrophils have helped the researchers identify additional elements of how P. gingivalis operates to subvert the immune system.
They found that the TLR2-C5aR crosstalk leads to degradation of the protein MyD88, which normally helps clear infection. And in a separate pathway from MyD88, they discovered that P. gingivalis activates the enzyme PI3K through C5aR-TLR2 crosstalk, promoting inflammation and inhibiting neutrophils' ability to phagocytose, or "eat," invading bacteria.
Inhibiting the activity of either PI3K or a molecule that acted upstream of PI3K called Mal restored the neutrophils' ability to clear P. gingivalis from the gums.
"P. gingivalis uses this connection between C5aR and TLR2 to disarm and dissociate the MyD88 pathway, which normally protects the host from infection, from the proinflammatory and immune-evasive pathway mediated by Mal and PI3K," Dr. Hajishengallis explained.
The research was supported by the National Institutes of Health, European Commission, and Medical Research Council.